Software Architecture
|
This document is currently undergoing revision. Please check back for updates, or contact us to get the most recent version. |
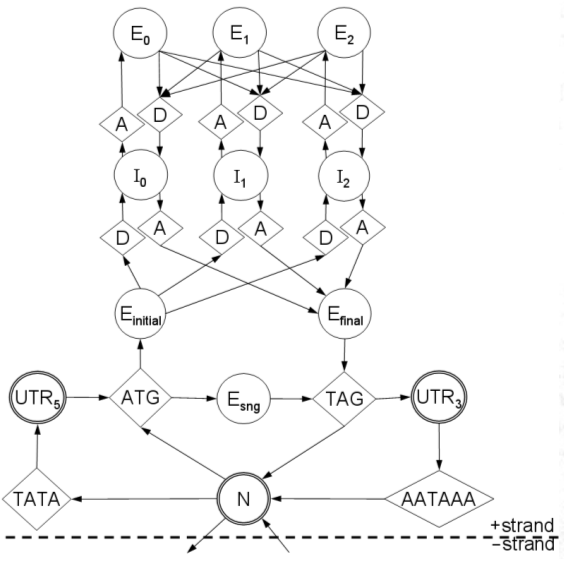
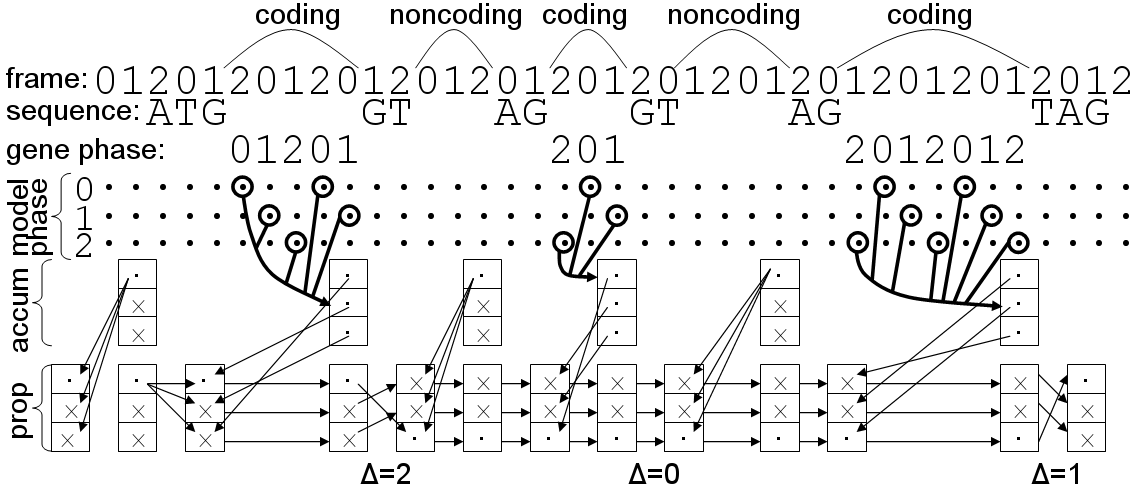
1. load sequence and model parameters.We will go through this line-by-line.
2. instantiate anchor signals at left terminus.
3. foreach base in the sequence, left-to-right:
4. foreach signal sensor:
5. if a signal occurs here:
6. instantiate a new signal S.
7. link S back to predecessors in appropriate queues.
8. enqueue S in all appropriate queues.
9. if a stop codon occurs here:
10. terminate this reading frame.
11. foreach signal queue:
12. propagate the queue's accumulator up to this point.
13. instantiate anchor signals at right terminus.
14. select highest-scoring right-terminus anchor R.
15. trace back from R to the left terminus to get optimal parse.
16. generate GFF from the optimal parse.
| Class |
Superclass | Description |
| Accumulator | Propagator |
a Propagator which stores an update delta to be added to all
the propagators in a queue |
| Alphabet |
a set of Symbols |
|
| AminoAlphabet |
Alphabet |
{*ARNDCQEGHILKMFPSTWYV} |
| ContentSensor | evaluates variable-length features of a parse (exons,
introns, etc.), one base at a time |
|
| DiscreteDistribution | maps integers to (log) probabilities |
|
| DnaAlphabet |
Alphabet |
{ACGNT} |
| EdgeFactory | manufactures Edge objects, or descendents of Edge (useful for
adding your own attributes to Edges through subclassing) |
|
| Edge | an edge connecting two Signals in a ParseGraph; abstract
base class |
|
| EmpiricalDistribution | DiscreteDistribution |
a DiscreteDistribution represented as a table of observed
frequencies |
| Fast3PMC | ContentSensor |
currently not in use |
| FastMarkovChain | ContentSensor |
currently not in use |
| GarbageCollector | a mark-and-sweep garbage collector for Signals |
|
| GarbageIgnorer |
GarbageCollector |
used to disable garbage collection |
| GeometricDistribution | DiscreteDistribution |
implements a geometric distribution,
P[length]=q*(1-q)^(length-1) where q=1/length |
| GffReader | builds a ParseGraph from the contents of a GFF file |
|
| IntronQueue | SignalQueue |
a type of SignalQueue which remembers only the best signal in
each phase |
| IntronQueueIterator |
TigrIterator <Signal*> |
allows iteration through the elements of an
IntronQueue's main queue (but not its holding queue) |
| IsochoreFile | loads a *.iso file and returns the appropriate
TigrConfigFile, given the actual GC content |
|
| LowercaseDnaAlphabet |
Alphabet |
{acgnt} |
| MarkovChainCompiler | currently not in use |
|
| MarkovChain | ContentSensor |
evaluates variable-length features of a parse using an Nth
order Markov Chain (and enforcing a min. sample size, like in IMMs) |
| MddTree | SignalSensor |
represents the tree used in MDD |
| ModelBuilder | builds a signal- or content-sensor from a set of training
sequences |
|
| NoncodingQueue | SignalQueue |
a type of SignalQueue which remembers only the best signal
(phase-agnostic) |
| NonPhasedEdge |
Edge |
an Edge representing a UTR or intergenic
segment, with a single phase-agnostic score |
| NthOrderStringIterator | generates all strings of length N, for installing
pseudocounts when training a Markov chain |
|
| ParseGraph | a set of Signals and Edges connecting those Signals into
valid parses |
|
| Partition | represents a positional test at an interior node in an MDD
tree |
|
| PhasedEdge | Edge | an Edge representing an exon or intron, with 3 phase-specific scores |
| Propagator | propagates the score of a path (in 3 phases) through a signal
at a specific point in the substrate sequence |
|
| ScoreAnalyzer | generates a precision-recall curve from a set of scores for
positive and negative examples |
|
| Sequence |
an array of Symbols drawn from an Alphabet |
|
| Signal | represents a fixed-length feature in a parse (splice site,
start-/stop-codon, promoter, poly-A signal) |
|
| SignalQueue | stores signals of a specific type that are still eligible to
participate in a parse at some par7ticular point later in the sequence |
|
| SignalSensor | a model that evaluates fixed-length features of a parse
(Signals) |
|
| SignalTypeProperties | consilidates knowledge about the properties of each signal
type, so that additional signal types can be added to the gene-finder
in the future |
|
| SinglePhaseComparator |
TigrComparator <Signal*> |
compares the propagated inductive scores of two
Signals in a given phase, accounting for accumulated lengths |
| Symbol |
a letter in an Alphabet |
|
| TataCapModel | SignalSensor |
a SignalSensor for TataCapSignals |
| TataCapSignal | Signal |
a Signal consisting of a TATA-box followed after a short
distance by a CAP-site |
| ThreePeriodicMarkovChain | ContentSensor |
a MarkovChain that models the three coding phases separately |
| TigrChi2IndepTest |
implements a chi-squared test for independence |
|
| TigrConfigFile |
reads and stores a set of named parameters from
a file |
|
| TigrFastaReader |
reads a fasta file |
|
| GeneZilla | encapsulates the entire gene-finder, so it can be embedded
within other projects |
|
| TopologyLoader | loads and parses the topology definition file |
|
| TrainingSequence | Sequence |
extends class Sequence with a "boost count" for implementing
boosting during training |
| Transitions | stores Transition probabilities |
|
| TreeNode | a node in an MddTree |
|
| WAM | SignalSensor |
a SignalSensor which is like a WMM but has a Markov chain at
each position in the matrix |
| WMM | SignalSensor |
a position-specific weight matrix |
| WWAM |
SignalSensor |
a WAM in which Markov chain probabilities were
estimated from pooled samples during training |
| Type | Values |
| Strand | PLUS_STRAND=FORWARD_STRAND, MINUS_STRAND=REVERSE_STRAND, EITHER_STRAND=NO_STRAND |
| SignalType | ATG, TAG, GT, AG, PROM, POLYA, NEG_ATG, NEG_TAG, NEG_GT,
NEG_AG, NEG_PROM, NEG_POLYA, NO_SIGNAL_TYPE |
| ModelType | WMM_MODEL, WAM_MODEL, WWAM_MODEL, THREE_PERIODIC,
MARKOV_CHAIN, FAST_MC, FAST_3PMC, IMM_MODEL, CODON_BIAS, MDD,
SIGNAL_MODEL, HMM_MODEL, NONSTATIONARY_MC |
| DistributionType |
EMPIRICAL_DISTRIBUTION, GEOMETRIC_DISTRIBUTION |
| Direction |
DIR_LEFT, DIR_RIGHT |