the
eukaryotic gene finder formerly known as

About GeneZilla
GeneZilla is a state-of-the-art gene finder based
on the
Generalized Hidden Markov Model framework, similar to Genscan and
Genie.
It is highly reconfigurable and includes software for retraining by the
end-user. It is
written in highly optimized C++. The run time and memory requirements
are linear in the sequence length, and are in general much better than
those of competing systems, due to GeneZilla's novel decoding
algorithm. Graph-theoretic representations of
the high scoring open reading frames are provided, allowing for
exploration of sub-optimal gene models. It utilizes Interpolated Markov
Models (IMMs), Maximal Dependence Decomposition (MDD), and includes
states for signal peptides, branch points, TATA boxes, CAP sites, and
will soon model CpG islands as well.
Accuracy
Results on 800 Arabidopsis
thaliana genes are shown below:
|
Nucleotide
|
Exon
|
Gene
|
|
Sn
|
Sp
|
Acc
|
Sn
|
Sp
|
Acc
|
 |
95%
|
98%
|
96%
|
77%
|
81%
|
43%
|
Genscan+
(trained for Arabidopsis)
|
93%
|
99%
|
95%
|
75%
|
82%
|
35%
|
Genscan
(trained for human)
|
69%
|
98%
|
80%
|
22%
|
43%
|
19%
|
Unveil
(a pure HMM trained for A. thaliana)
|
95%
|
84%
|
87%
|
40%
|
36%
|
7%
|
Note that the training and test sets were disjoint for all results
reported on this page. On the "standard" Bursett/Guigo test set
of 558 vertebrate genes GeneZilla performed very similarly to Genscan
|
nucleotide
|
splice site
|
start/stop codon
|
exons
|
exact
|
|
Sn
|
Sp
|
F
|
Sn
|
Sp
|
Sn
|
Sp
|
Sn
|
Sp
|
F
|
genes
|
|
95%
|
96%
|
96%
|
89%
|
87%
|
82%
|
79%
|
82%
|
80%
|
81%
|
51%
|
Genscan
|
96%
|
97%
|
97%
|
90%
|
89%
|
72%
|
89%
|
81%
|
84%
|
82%
|
43%
|
Efficiency
Time and memory usage on a 922 Kb
Aspergillus
fumigatus contig are shown below:
|
Memory (Mb)
|
Time
(min:sec)
|
|
29
|
1:28
|
Genscan
|
445
|
2:57
|
These results demonstrate that
GeneZilla is extremely memory efficient while also achieve higher
speeds than Genscan. GeneZilla has successfully
processed contigs as large as 2 Mb on an ordinary laptop
computer. The excellent space efficiency of GeneZilla allows it
to be used as a component of more sophisticated systems such as
comparative gene finders, while leaving more memory for the comparative
analyses.
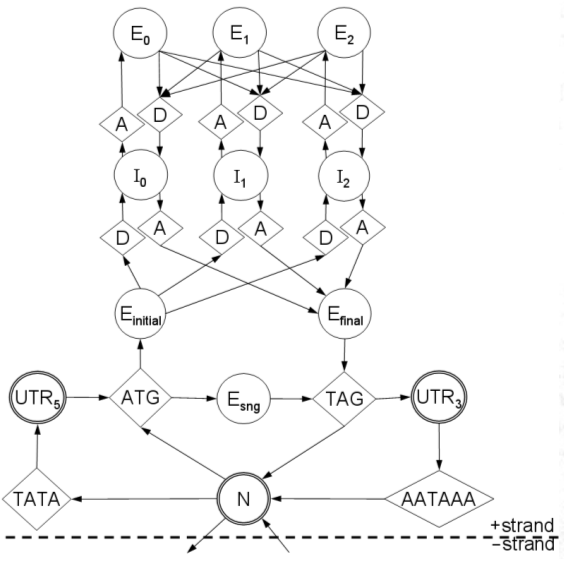
Architecture
GeneZilla's state-transition diagram is
essentially the same as that of Genscan. GeneZilla has the
ability to model different types of exons (i.e.,
initial/internal/final/single) using different content sensors, unlike
most GHMM-based gene finders. The state diagram shown below
models only forward strand genes; reverse-strand genes are handled by a
mirror-image of this model, and are not permitted to overlap with
forward-strand precitions.
Not shown in this diagram are the signal
peptide, CAP site, and branch point models. GeneZilla will also
soon possess an (optional) CpG island state upstream from the start
codon for use when applied to vertebrate genomes.
More information about GeneZilla's software architecture can be found here.
GeneZilla (formerly TIGRscan) is briefly described in:
Majoros W, et al. (2004) TIGRscan and
GlimmerHMM: two open-source ab initio
eukaryotic gene finders, Bioinformatics
20, 2878-2879.
The novel decoding algorithm used by GeneZilla is described in:
Majoros W. et al. (2005) Efficient decoding
algorithms for generalized hidden Markov model gene finders, BMC Bioinformatics 5:616.
Downloads
GeneZilla is available for download
as OSI
Certified Open Source Software under the Artistic
License. Pre-trained model files are
also available.